


¿Qué es la osteogénesis imperfecta?
La osteogénesis imperfecta es una enfermedad del metabolismo óseo causada por alteraciones en la producción del colágeno tipo I, proteína fundamental en la constitución de los huesos.
Esta enfermedad provoca que los huesos sean frágiles y presentan un riesgo mayor de fracturarse, tras un mínimo golpe e incluso espontáneamente. Se dice que las personas que la padecen poseen “huesos de cristal”.
¿Cuál es la causa que lo provoca?
La osteogénesis imperfecta es una enfermedad hereditaria producida por un defecto en el cromosoma 7 (gen COL1A1) o en el cromosoma 17 (COL1A2) aunque pueden producirse también por mutaciones no hereditarias de estos genes, o de otros genes, como ocurre en las formal letales de la OI asociadas a mutaciones en los genes CRTAP y LEPRE1.
La mutación de estos genes causa que ocurra la sustitución de una glicina en la molécula de procolágeno, produciendo así una formación de cadenas de colágeno anormales. Existen varias formas de esta enfermedad (tipo I hasta tipo VII).
Es una enfermedad congénita (presente desde el nacimiento) producida por alteraciones en la cantidad o calidad de formación del colágeno y que provoca que la formación de hueso sea defectuosa y de mayor fragilidad secundario a una producción insuficiente de osteoide, alterando las capacidades de remodelación de hueso.
¿Cuáles son los síntomas de la osteogénesis imperfecta?
La manifestación más evidente es la fragilidad ósea que provoca la fractura frecuente y anormal de los huesos. Estas fracturas podrán sanar normalmente de manera inicial, pero una característica particular es que posteriormente no presentan remodelación. Incluso, una de las manifestaciones iniciales de esta enfermedad es una fractura por avulsión de la apófisis del olécranon.
Existen también otros síntomas asociados dependiendo del subtipo de osteogénesis que se padezca entre los que destacan:
- Escleróticas azules (coloración azul en la parte blanca de los ojos)
- Problemas de audición y sordera en hasta 50% de los casos
- Deformidad de las caderas (coxa vara), que produce alteraciones en la marcha
- Deformidad de las piernas (tibias con deformidad "en sable")
- Problemas en los dientes (dientes frágiles y coloración amarilla)
- Estatura baja
- Cifosis o escoliosis (curvatura anormal de la columna)
- Escaso desarrollo muscular
- Laxitud ligamentaria
- Problemas respiratorios
- Problemas cardiacos, predominantemente prolapso de la válvula mitral, y regurgitación aórtica
¿Qué tipos existen?
Existen varios tipos de osteogénesis imperfecta con diferente grado de severidad. El tipo dependerá de la mutación subyacente; los casos con un patrón de herencia “dominante” podrán ser leves o severos, y cursarán con los tipos I a IV de esta enfermedad, mientras que aquellos con un patrón “recesivo” darán lugar a los tipos VI y VII.
Los más frecuentes son:
- Tipo I (leve o enfermedad de Ekman Lobstein): es la forma más leve de la enfermedad, presentan estatura normal, escasa o nula deformidad en los huesos, sordera en el 50% de los casos, escleras azules y a veces alteraciones en los dientes.
- Tipo II (letal perinatal o Enfermedad de Vrolik): afecta al bebé dentro del útero, siendo mortal en la mayoría de los casos, con huesos deformados y fracturados.
- Tipo III (deformante progresiva): es la forma sobrevivible más severa, presentando deformidad ósea progresiva, generalmente con deformidades leves y fracturas al nacimiento, escleróticas azules, alteraciones en la dentición, sordera frecuente y estatura muy corta.
- Tipo IV (moderada): forma de severidad moderada, con escleróticas normales, deformidad ósea moderada o leve, fracturas vertebrales, y estatura variable, pero sin alteraciones en la audición. La alteración en la dentición es también frecuente.
¿Cómo se puede detectar?
El diagnóstico de esta enfermedad se basa en la recolección e identificación de una historia familiar positiva con antecedentes para osteogénesis imperfecta, aunado a hallazgos clínicos e imagenológicos.
Se suele utilizar la densitometría para medir la masa ósea. La realización de densitometrías de forma periódica permite valorar la evolución de la enfermedad y determinar la probabilidad de fracturas.
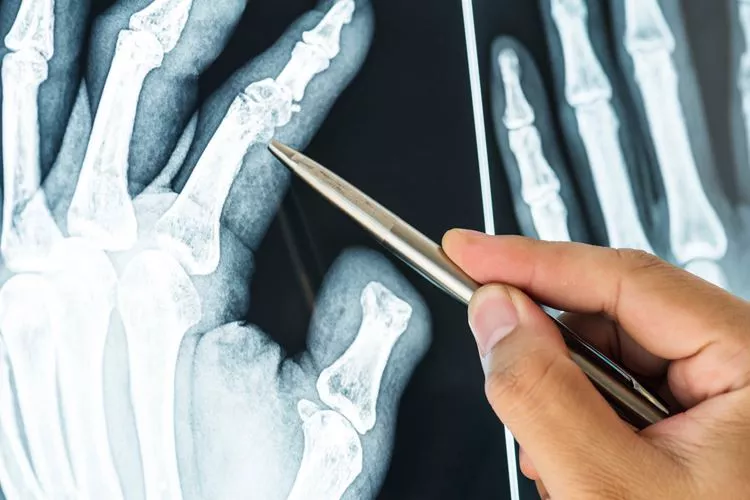
En una radiografía simple, se podrán identificar las siguientes características que elevan la sospecha ante un caso de osteogénesis imperfecta: corticales delgadas, osteopenia generalizada, tibias “en sable”, y deformidad en las caderas (coxa vara).
Se debe realizar asimismo una audiometría para comprobar si existen problemas de audición.
Durante el embarazo se puede realizar una biopsia de vellosidades coriónicas para saber si el feto padece la enfermedad.
¿Cuál es el tratamiento recomendado?
No existe tratamiento curativo en la actualidad.
El tratamiento actual es paliativo y consiste en:
- Intervenciones quirúrgicas para reforzar los huesos largos del organismo introduciendo clavos dentro del hueso (cirugía de Rodding).
- Manejo por medio de ortesis externas para evitar la deformidad de las extremidades, y reducir el riesgo de fracturas.
- Utilización de los Bisfosfonatos y Pamidronato para mejorar la densidad de los huesos, aunque su uso no ha mostrado beneficios en la corrección de la escoliosis, y su uso continuo puede producir un cierre fisario temprano.
- Ejercicio físico de bajo impacto (natación) para potenciar el desarrollo muscular.
En caso de que se presente una fractura, se ofrecerá el tratamiento de acuerdo al tiempo específico y a la presentación de la fractura.


Fernando Martínez Sáez
Redactor y divulgador de temas científicos, médicos y sanitarios. Miembro de la Asociación Nacional de Informadores de Salud.
Autor originalDr. Jorge Valenzuela Flores
Especialista en Ortopedia y Traumatología y licenciado en medicina por el Instituto Tecnológico y de Estudios Superiores de Monterrey (TEC).
Revisor clínico